

Actinium Pharmaceuticals (NYSEMKT: ATNM) had a very successful showing at the American Society of Hematology (ASH) annual meeting held over the past few days in Orlando, FL. The highlight of the event for the company was a poster presentation of Phase 1 clinical data with Actimab-A, the company’s CD33 targeting alpha particle immunotherapy for the treatment of acute myeloid leukemia (AML). Below is a quick background on Actimab-A and a summary of the poster data presented at ASH.
Quick Background On Actimab-A
Actimab-A a next-generation monoclonal antibody linked to radioactive actinium-225 (Ac-225). Actimab-A consists of lintuzumab, a humanized monoclonal antibody that targets the myeloid progenitor cell protein CD33. CD33 is a transmembrane receptor leukocyte antigen primarily expressed on cancer cells of myeloid lineage, although it can also be found on some lymphoid cells. Lintuzumab was originally developed by Seattle Genetics with ties back to Memorial Sloan Kettering. Seattle Genetics attempted to develop lintuzumab as a treatment for AML and other myeloproliferative diseases, but a Phase 2b clinical trial failed to demonstrate an increase in overall survival and the program was discounted in 2010 (source).
The concept of Actimab-A holds significant scientific merit. Lintuzumab was proven to be safe and tolerable in the Seattle Genetics clinical work and CD33 is a commercially validated target. Pfizer’s Mylotarg® (gemtuzumab ozogamicin) was an anti-CD33 murine antibody conjugated to the chemotherapeutic agent, calicheamicin hydrazide. Mylotarg® demonstrated success in mid-stage clinical trials (Hamann PR, et al., 2001), earning Pfizer accelerated approval from the U.S. FDA for the drug in 2000; however, safety concerns and lackluster post-approval trials ultimately led to the drug being withdrawn from the market in 2010.
Lintuzumab by itself has proven to be ineffective; but, linked to the short-ranged (50-80 µm), high-energy (~100 keV/µm) alpha particle–emitting radioactive isotope Ac-225, Actimab-A should see a powerful increase in cancer cell-killing effect. Actimab-A is a next-generation product to Actinium’s previous lintuzumab-linked alpha particle emitter, Bismab-A. Bismab-A utilized bismuth-213 and demonstrated positive results in a Phase 1/2 clinical trial – complete response rates in the 30% range, superior to that of Mylotarg® – but the drug was hampered by the short half-life of bismuth-213 (~45 minutes) and lacked commercial viability due to the high cost of goods and complex on-site preparation. Actinium-225 has a half-life of approximately 10 days and should offer significantly improved costs with centralized manufacturing.
Phase 1 Data At ASH
Actinium Pharma is currently in a multi-center Phase 1/2 clinical trial (NCT01756677) with Actimab-A as a first-line treatment for AML patients over the age of 60 who are not suitable for standard induction chemotherapy. This is most likely because these patients have antecedent hematologic disorder, unfavorable cytogenetic or molecular abnormalities, and significant comorbidities that likely makes standard induction intolerable to these patients. The trial is being conducted at five centers in the U.S., Memorial Sloan Kettering, Fred Hutchinson, Johns Hopkins, Baylor Cancer Center, and the University of Pennsylvania.
The goal of the Phase 1 part of this study is to find the highest tolerable dose of Actimab-A that can be given with cytarabine, a commonly used anti-cancer drug, to patients with AML. At ASH, Actinium presented data from fourteen patients treated to date (median age: 77 years). Actimab-A was given at 0.5 (n=3), 1 (n=6), 1.5 (n=3), or 2 (n=2) μCi/kg/fraction. Up to four cycles of low-dose cytarabine were administered. Dose-limiting toxicities were seen in one patient at 1 µCi/kg/fraction who had grade 4 thrombocytopenia with marrow aplasia for more than 6 weeks following therapy. Hematologic toxicities included grade 4 neutropenia (n=4) and thrombocytopenia (n=6). Grade 3/4 non-hematologic toxicities included febrile neutropenia (n=7), pneumonia (n=4), bacteremia (n=1), cellulitis (n=1), transient creatinine increase (n=1), hypokalemia (n=1), rectal hemorrhage (n=1), and generalized weakness (n=2).
Eight of 11 patients (73%) evaluated after one cycle had bone marrow blast reductions (mean reduction, 72%; range, 34-100%). Seven (64%) had blast reductions of at least 50%.
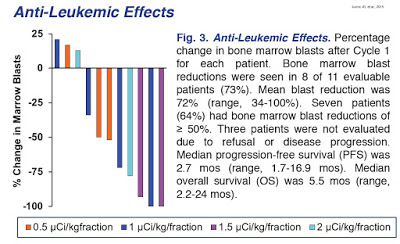
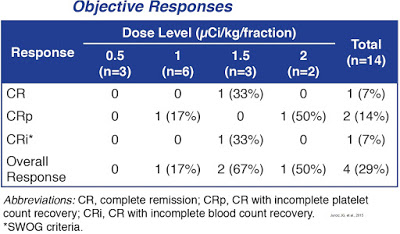
Objective responses (1 CR, 1 CRp, 2 CRi) were seen in four of the 14 patients (29%) after one cycle of therapy. Responses were seen only at doses ≥ 1 µCi/kg/fraction (4 of 11 patients, 36%). Median progression-free survival (PFS) was 2.7 months (range, 1.7-16.9 months). Median overall survival (OS) was 5.5 months (range, 2.2-24 months).
 Conclusion
Conclusion
Data at ASH demonstrates the clear anti-leukemia effect of Actimab-A in elderly patients with AML not eligible for standard induction therapy. Based on previous and newly-presented data, Actimab-A is tolerable and effective at 1-2 μCi/kg/fraction, and can be safely combined with low-dose cytarabine to produce remission in older patients with untreated AML. The company is continuing with the dose escalation Phase 1 part of the study designed to define the maximum tolerated dose. Once complete, Actinium will enroll additional patients in the Phase 2 portion of the trial, which is designed to determine the response rate, progression-free survival, and overall survival in this difficult to treat population. The trial is designed to enroll a total of 72 patients in total.
I believe the target market for Actimab-A is meaningful. Approximately two-thirds of the AML population is over the age of 60 years old and half of these patients are not eligible for the current standard of care due to overlapping toxicities with contaminant medications, existing co-morbidities, and/or physician expectations on the patient’s ability to tolerate standard “7+3” induction. Complete remission to low-dose cytarabine is typically only seen in ~20% of patients (Burnett AK, et al., 2007), and Phase 1 data presented at ASH looks superior to this standard.
In December 2014, Actimab-A was granted Orphan Drug status by the U.S. FDA (see release). I estimate the patient population in the U.S. is approximately 6,500 individuals. The opportunity in Europe is another 7,500. Assuming a cost of approximately $65,000 per treatment in the U.S. and Japan and $42,500 in the EU (management guidance), the peak opportunity for Actimab-A is easily $750 million. With 33% market share, Actimab-A looks to have sales in the $250 million range.